Analysis Overview
Combocat provides a streamlined pipeline for analyzing drug combination screening data, integrating experimental and computational workflows into a single R package. This section outlines the primary steps for downloading, installing, and using the package to analyze your data.
Install the R package
![]()
The combocat R package is hosted on GitHub and can be installed directly using devtools.
Install required dependencies
Ensure you have R installed on your system, and install the devtools package if you don’t already have it:
install.packages("devtools")Install and load combocat
#Install from GitHub
devtools::install_github("wcwr/combocat")
#Load package
library(combocat)Several dependencies will be installed along with the package. Notably, dplyr, which is the tidyverse grammar framework that combocat adopts throughout for its user-friendly approach to data manipulation and analysis.
Access documentation of functions
The package includes documentation for each function. Use the help() or ?? commands to understand specific functions:
??cc_map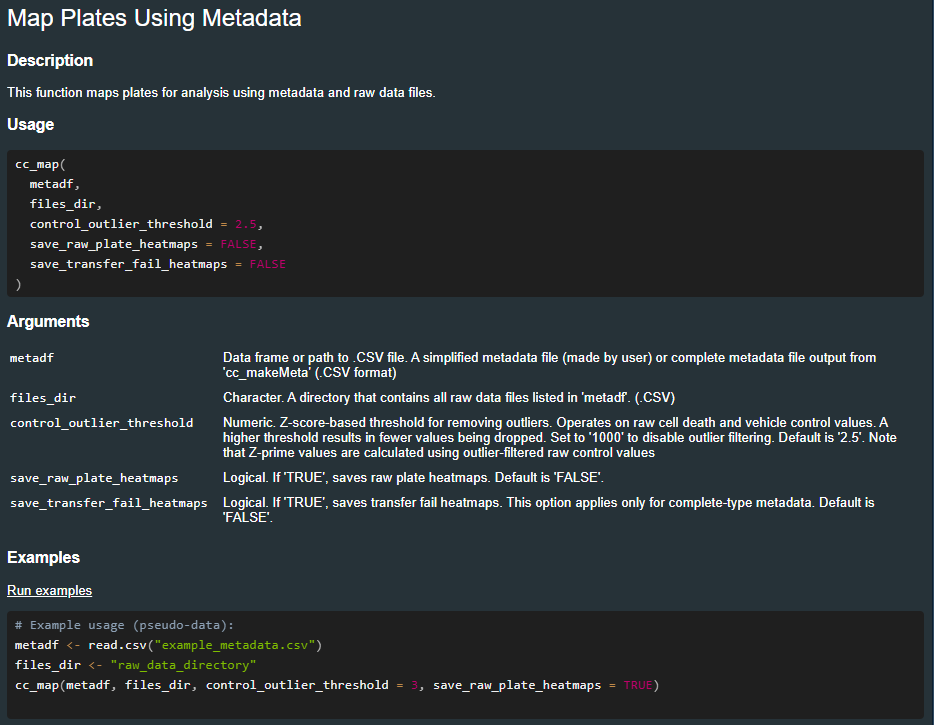
Workflow Overview
The workflow is designed to be straightforward, and is packaged into a handful of streamlined functions:

Mapping associates each raw value with its corresponding position in the combination matrix
Normalization uses within-plate controls to represent the data as a function of % cell death
Single-agents are fit with dose-response curves to serve as a basis for synergy calculations
Synergy is calculated using the Bliss independence model

Each core function is designed to work regardless of dense or sparse mode data
Metadata
Metadata forms the backbone of any Combocat analysis by linking raw data files to experimental conditions like drug names, doses, and file names. Properly structured metadata facilitates correct analysis–mainly the mapping stage.
Generating the (dense mode) metadata file is easy 👌
You will need:
drug1_nameanddrug2_name(which must not contain underscores_)drug1_concanddrug2_concwhich list the 10 doses used for each drugunitswhich must be the same for both drugssample_name- the name of a cell line/sample (must be unique for each combination)filename: the name of the file
For example, the metadata for a single combination looks like this:
You can download this (File ➝ Download) to follow along. Be sure to choose .csv
Rename the file to meta_dense.csv so the filename matches!
Under the hood, the cc_map function will expand this to all pairwise combinations of doses.
If more combinations are used, their info can be added to the metadata file. There will only be 1 metadata file per experiment.